(www.genome.ad.jp)
(www.genome.ad.jp/dbget/dbget.links.html)
国際共同研究として始まったゲノムプロジェクトは、いまや国際戦争である。各国政府が投入する膨大なゲノム関連予算に対し、民間企業もさらに大規模な投資を行い、早く情報をおさえ知的所有権を獲得するために、ゲノムのシークエンシング(塩基配列決定)競争に駆り立てられている。しかしながら、その実体は必ずしもサイエンスとしての健全な競争ではなく、バブル期の競争的な投資に似ている。ゲノムには情報がある、その情報には価値がある、といった漠然とした思惑で、ともかく突っ走っている。また、科学者が使う言葉の意味を正確に伝えずに、ゲノムの情報を解読した、情報をデータベース化した、といった発表がなされ、マネーゲームが行われている。このような情報は information ではなく、disinformation である。
そもそもゲノムプロジェクトとは、ヒトの全遺伝情報であるゲノムを構成する30億文字分のDNA塩基配列(A, C, G, T の並び)を決定するプロジェクトである。同時にバクテリアから高等動植物まで、様々な生物種でも全塩基配列の決定が行われている。当初は15年かけて2005年までにヒトゲノムを決定する予定であったが、その後プロジェクトの順調な進展により2年早められて2003年までに達成できるとされていた。ところが昨年春以来セレラ・ゲノミックス社(www.celera.com)の挑発的な言動により、2001年には、いや2000年春には、といった具合に達成時期がどんどん早まっている。セレラは日本が10年かけてやり遂げる予定であったイネゲノムを、6週間あれば終了できるとも宣言している。
もっともこのような終了宣言は、何をもってシークエンシング完了と定義するかが曖昧なことを利用した、単なる言葉のあそびでしかない。ビットの読者なら自分でプログラムを書いた経験のある方は多いだろう。90%のコードを書いてプログラムを大ざっぱに仕上げるのは2〜3日でできても、バグをとって100%仕上げるためにはさらに1ヶ月かかるといったことは、ごくあたりまえの話である。ゲノムのシークエンシングにしても、90%読むのは6週間でできても、100%に到達するには何年もかかり得る。90%できあがったプログラムがほとんど役に立たないように、90%の精度でゲノムを読んでみても、有用な情報はあまり得られないと思われる。
昨年12月に初の多細胞生物として線虫のゲノムが決定されたと発表された。ところがこの線虫ゲノムはまだ虫食いだらけで、本当の意味ではシークエンシングは終了していないのだが、当初からの公約で1998年中に終了宣言をすることが必要だったのだろう。まあ線虫の場合は遺伝子がほとんど分かっているので実質的に終了した(virtually complete)と言ってもいい。しかし、このような曖昧な言い方をアカデミック側がしているため、セレラの極端なやり方も非難できないわけである。
もう1つの言葉のあそびに「解読」がある。ゲノムを解読したと言われれば、一般社会の常識として、そこに何が書いてあるかが分かったと思うだろう。しかしながら、生物学者が言うゲノムの解読とはあくまでもゲノムを A, C, G, T の文字列の並びとして読みとることである。英語を全く知らない人でも英語の文章を与えられると、その意味は分からなくても、26文字のアルファベットが機械的にどんな順番で並んでいるかは分かる。ゲノムの解読とは、とくにヒトゲノムの解読とは所詮そのようなもので、ゲノムに書かれた生物的な意味が分かることとは別問題である。現にこれまで全塩基配列が決定され公表された20種以上の生物種において、どんなはたらきをしているかまで理解できた遺伝子の数は平均すると半分以下しかない。ヒトゲノムにおいては個々の遺伝子がどこにあるかさえはっきり推定できない場合が多く、機能まで到達できる遺伝子の割合はもっと低くなるであろう。
一方、実はここに情報科学としてのゲノムの面白さがある。真の意味でゲノムを解読することは、まだどの生物種においてもできていない。ゲノムの配列情報から生物的な意味を解釈することは、基本的に情報科学の問題である。そしてもしゲノムが生命の設計図なら、生命のシステムをコンピュータの中に再構築できるはずである。ゲノムプロジェクトがもたらす配列情報は、公共的なデータベースとしてインターネット上で公開されており、ゲノムの解読は専門の研究者だけでなく、誰でも同じ土俵の上で取り組むことができる。21世紀の生物学の大発見は、インターネットを使ってゲノムを解析した情報科学者がもたらすことも充分に考えられる。本稿ではそのような意欲のある人のために、我々が提供するゲノムネット [1][2] を中心に、ゲノム関連のインターネットリソースを紹介する。
|
|
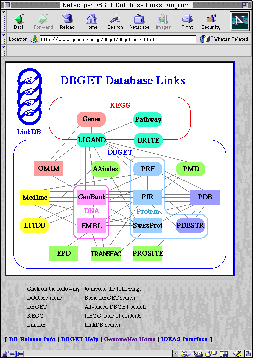
| 図1.ゲノムネットのホームページ (www.genome.ad.jp) | 図2.DBGET/LinkDB システム (www.genome.ad.jp/dbget/dbget.links.html) |
|---|
| 機 関 | アドレス | 主要検索システム | 作成データベース |
|---|---|---|---|
| ゲノムネット (京都大学化学研究所) | www.genome.ad.jp | DBGET/LinkDB | KEGG |
| NCBI (米国バイオテクノロジー情報センター) | www.ncbi.nlm.nih.gov | Entrez, BLAST | Medline, GenBank |
| EBI (欧州バイオインフォーマティクス研究所) | www.ebi.ac.uk | SRS | EMBL, SWISS-PROT |
| SIB (スイスバイオインフォーマティクス研究所) | www.expasy.ch | SRS | SWISS-PROT |
WWW では世界中に存在するコンピュータ上の各ファイルをホスト名とマシン名のペアで指定する。例えば、KEGG のホームページは
www.genome.ad.jp/kegg/kegg.htmlであり、ここから筆者のホームページへリンクがつけられている。
www.genome.ad.jp/kegg/kegg.html → kanehisa.kuicr.kyoto-u.ac.jp/index.html同様に、DBGET/LinkDB などの統合システムでは世界中に存在するデータベース中の各エントリーをデータベース名とエントリー名のペアで指定する。例えば、米国 DNA データベース GenBank の乳がん遺伝子 BRCA1 の配列情報は
GenBank:HSU14680であり、これを発表した論文は、医学・生物学関連文献データベース Medline へのリンクとして表現されている。
GenBank:HSU14680 → Medline:95025896さらに、 KEGG ではヒトの遺伝子とマウスの遺伝子の類似性
H.sapiens:BRCA1 → M.musculus:Brca1あるいは乳がん遺伝子と別の遺伝子との関連
H.sapiens:BRCA1 → H.sapiens:RAD51といった生物学的な関係もリンクとして表現されている。
| データの内容 | データベース名 | メディア |
|---|---|---|
| 塩基配列 | GenBank(DDBJ含む), EMBL | テキスト |
| アミノ酸配列 | SWISS-PROT, PIR, PRF, PDBSTR | テキスト |
| 立体構造 | PBD | テキスト、三次元グラフィックス |
| 配列モチーフ | EPD, TRANSFAC, PROSITE BLOCKS, ProDom, PRINTS, Pfam | テキスト、三次元グラフィックス |
| 酵素 | LIGAND/ENZYME | テキスト |
| 化合物 | LIGAND/COMPOUND | テキスト、イメージ、
二次元グラフィックス |
| 化学反応 | LIGAND/REACTION | テキスト |
| 分子間相互作用 | BRITE | テキスト |
| パスウェイ | KEGG/PATHWAY | テキスト、イメージ |
| 遺伝子 | KEGG/GENES | テキスト |
| ゲノムマップ | KEGG/GENOME | テキスト、イメージ、
Javaアプレット |
| 発現マップ | KEGG/EXPRESSION | テキスト、イメージ、
Javaアプレット |
| 遺伝病 | OMIN | テキスト |
| アミノ酸変異 | PMD | テキスト |
| アミノ酸指標 | AAindex | テキスト |
| 文献 | Medline, LITDB | テキスト |
| リンク情報 | LinkDB | テキスト |
表2にゲノムネットの DBGET/LinkDB システムに組み込まれたデータベースの一覧を示した。ゲノム情報の最も基礎となるDNA塩基配列については、NCBI (National Center for Biotechnology Information) の GenBank データベース、EBI (European Bioinformatics Institute) の EMBL データベース、それに国立遺伝学研究所の DDBJ データベースが、国際協力の下にデータベース化を行っている。塩基配列以外の情報については、米国では NCBI が 文献情報データベース Medline (PubMed) を自由にアクセスできるようにし、各出版社にある論文の全テキスト情報とリンクを張りめぐらせて、膨大な情報ネットワークを作りつつある。また、NCBI は GenBank を PubMed だけでなく、アミノ酸配列、立体構造、ゲノムマップなどの情報と統合した Entrez システムを提供している。ヨーロッパでは EBI と SIB (Swiss Institute of Bioinformatics) が共同で SWISS-PROT アミノ酸配列データベースの構築を行っている。ヨーロッパには Entrez のような一極化したサービスはないが、EBI が開発した SRS システムが各地に配布され、分散サービス体制となっている。
ゲノムネットの最大の売り物はリンクの概念を生物的なつながりに拡張した KEGG システムである。そもそも生物の機能とは単独の遺伝子や単独のタンパク質に帰属できるものではなく、多数の遺伝子および分子の相互作用により実現されている。KEGG では生物機能を遺伝子または分子のネットワークとして表現した PATHWAY データベースと、各生物種のゲノムプロジェクトから決定される遺伝子カタログを集積した GENES データベースを中心に、生命システムの配線図を明らかにする研究が進められている。KEGG については次回に詳しい解説があるので、詳細はここでは省略する。
ゲノムネットのもう1つの特徴はリンク情報をダイナミックに扱っている点である。いわゆる三段論法とは、2つのリンク A → B と B → C から新しいリンク A → C を導くことに相当する。このように複数のリンクを組み合わせて新しいリンクを導き出す機能、すなわち二項関係から演繹する機能があることから、DBGET/LinkDB システムや KEGG システムは簡単な演繹データベースシステムでもある。図2に DBGET データベースのリンク図が示されている。これは各データベースをノード、リンク情報が存在するデータベース間のつながりをエッジとしたグラフである。この場合の演繹機能とは、エッジが存在しないデータベース間でも、他のエッジをたどることによりリンクをつける機能である。グラフで推移閉包を求めることに相当する。DBGET/LinkDB システムでは重要な経路についてはあらかじめ計算してあるが、ダイナミックに計算する機能もあり、例えばユーザー独自のデータベースが SWISS-PROT だけにリンクされていれば、ゲノムネットデータベースのすべてに対してのリンクを求めることができる。
| 生物科学の問題 | 計算機科学の方法 | ||
|---|---|---|---|
| 類似性検索 | ペアワイズアライメント
ホモロジーサーチ マルチプルアライメント 系統樹解析 立体構造アライメント |
最適化アルゴリズム
・ダイナミックプログラミング(DP) ・シミュレーテッドアニーリング(SA) ・遺伝的アルゴリズム(GA) ・ホップフィールドネットワーク | |
| 構造・機能予測 | アブイニシオ法 | RNA二次構造予測
RNA立体構造モデリング タンパク質立体構造予測 | |
| 知識ベース法 | 配列モチーフ抽出
機能部位予測 細胞内局在部位予測 遺伝子コード領域予測 膜貫通領域予測 タンパク質二次構造予測 タンパク質立体構造予測 |
パターン認識・学習アルゴリズム
・判別分析 ・ニューラルネットワーク(ANN) ・隠れマルコフモデル(HMM) ・形式文法 | |
| 分類 | スーパーファミリー分類
フォールド分類 オーソログ・パラログ遺伝子分類 | クラスタリングアルゴリズム
・階層的クラスター解析 ・コホーネンネットワーク | |
| 内容 | プログラム名 | アドレス | ホモロジー検索 | BLAST FASTA |
www.blast.genome.ad.jp
www.fasta.genome.ad.jp |
モチーフ検索 | MOTIF TFSEARCH |
www.motif.genome.ad.jp
www.cbrc.jp/research/db/TFSEARCH.html |
マルチブルアライメント | CLUSTALW | www.clustalw.genome.ad.jp | 細胞内局在部位予測 | PSORT | psort.nibb.ac.jp | 膜貫通領域予測 | SOSUI | www.tuat.ac.jp/~mitaku/adv_sosui/ | 膜タンパク質予測 | TSEG | www.genome.ad.jp/SIT/tseg.html | 遺伝子領域予測 | GRAIL | grail.genome.ad.jp | 立体構造可視化 | PACKADE | pacade.genome.ad.jp/pdb_highligt.html | ネットワーク予測 | KEGG | www.genome.ad.jp/kegg/kegg3.html |
|---|
ゲノムネットでは表4に示した配列解釈ツールが利用可能である。世界中で標準的に使われているホモロジー検索システムである BLAST と FASTA、我々が開発したモチーフ検索システム MOTIF、マルチプルアライメントで標準的な CLUSTALW、そして TFSEARCH、PSORT、SOSUI をはじめ我が国の研究者が開発したユニークなツール群へはリンクとして、サービスが提供されている。BLAST、FASTA、MOTIF などの検索結果は DBGET/LinkDB システムに組み込まれ、関連する情報を様々なデータベースから取得することができる。実際、ホモロジー検索またはモチーフ検索の結果は、問い合わせ配列と関連した配列(近傍配列と呼ぶことがある)へのリンクの集まりであるとみなすことができ、ゲノムネットのリンク計算に組み込むことができる。