図1 ヒューマンゲノム計画
ヒューマンゲノム計画は、分子生物学の技術革新の流れと医学的価値への期待から、1980年代の終わりに始まった生命科学のビッグプロジェクトである。様々な生物ゲノムの全塩基配列という大量データの出現に対処するため、当初から新しい情報処理技術の開発と情報インフラストラクチャーの整備に重点が置かれ、計画の波及効果として生命科学全般の情報化を引き起こしつつある。ヒューマンゲノム計画がもたらす分子レベルの情報は、分子間相互作用の概念の下に、細胞レベル、個体レベル、生物種レベルの情報、そして自然界における生物界の情報をも明らかにしつつある。
「ヒューマンゲノム計画」の系譜をたどると、エンドウの交雑実験から遺伝子の概念を確立したメンデルを出発点とする遺伝学の流れと、細胞の顕微鏡観察から染色体に全遺伝情報であるゲノムが存在することを明らかにした細胞生物学の流れに到達する。遺伝子(gene)やゲノム(genome)という言葉ができたのは20世紀になってからであるが、19世紀に誕生した遺伝学と細胞生物学は現代生物学の流れを形成した。そして、両者を極言すると「はたらき」から探る生物学と、「かたち」から探る生物学の流れでもある。
20世紀後半の生物学は分子生物学全盛の時代であった。1953年に提唱されたワトソン・クリックのDNA二重らせんモデル [1] では、「かたち」すなわち塩基対の相補的な立体構造の中に、「はたらき」すなわち遺伝情報伝達のメカニズムが隠されていることを明らかにした。分子生物学は一見「かたち」から出発する学問のように思われるかもしれないが、一般の生物学者は特定の「はたらき」にまず興味の対象があり、それを司る分子の「かたち」を調べるために分子生物学の実験技術が用いられてきたのである。
ヒューマンゲノム計画は、後述するように実験技術の面では分子生物学の技術革新の結果として可能となったプロジェクトである。しかしながら、発想としてはそれまでの分子生物学とは逆であり、一次元的な「かたち」すなわちゲノムの全塩基配列を決定することに目標がある。これをもとに個々の遺伝子の「はたらき」を調べることが容易になるわけで、ゲノム計画はそのためのインフラストラクチャー作りと位置づけることもできる。本章ではヒューマンゲノム計画の概要と、ヒューマンゲノム計画がもたらしつつある新しい生物学、ニューバイオフィジックスについて概観する。
制限酵素の発見によりDNAを切ったりつないだりするすることができるようになり、特定のDNA断片を増幅するクローニング技術、長さの異なるDNA断片を分離する技術、それに基づくDNA塩基配列決定技術といった遺伝子工学の技術は、1970年代に確立した [2]。その結果、1980年代は遺伝子クローニング全盛の時代であった。特定の機能を司るタンパク質分子をコードする遺伝子を探すこと、とくにメッセンジャーRNAを探してcDNAクローニングとシーケンシングを行うことが一般的であった。さらに、そのcDNAをプローブとしてゲノムDNAの部分的シーケンシングを行い、エキソン・イントロン構造などが調べられた。
しかしながら、たまたま興味をもった特定の「はたらき」から出発し、個別に遺伝子クローニングを行うことで、例えばヒトがもつすべての遺伝子を調べあげることは不可能に近い。一方、ゲノムの中にはすべての遺伝子が含まれているのだから、ゲノムの塩基配列を端から端まで決めてしまい、そこから遺伝子を同定し機能を推定するアプローチがとれないだろうか。これが遺伝子クローニングからゲノム解析への発想の転換であった。個々の「はたらき」を知るためには、いずれにせよ個々の「かたち」を調べなければならないのだから、先にすべての「かたち」を決めておいた方が、最終的には効率がよいとの考えである。
もっとも、ヒトの全ゲノムをシーケンシングするためには、大きなDNAを取り扱う技術開発が必要であった。1980年代前半の技術でもウイルス、ミトコンドリア、クロロプラストなど小さな(105 塩基程度の)ゲノムの全塩基配列決定は可能であったが、1980年代の後半に YAC ベクター、パルスフィールド電気泳動法、PCR 法などの技術が確立して、ヒューマンゲノム計画が可能となったのである。
1990年代に入ってゲノム計画は本格化した。ウイルスのように宿主に依存するのではなく、単独で生きる生物として初めてのバクテリアゲノムが1995年に決定され [3]、翌年には初の真核生物として酵母のゲノムシーケンシングが終了した。ゲノムの全塩基配列が決定されると、そこから遺伝子領域を予測し(「1−2 タンパク質コード領域予測」参照)、遺伝子産物の機能を推定することは情報処理の問題となる。機能推定は類似配列がデータベース(「1−4 データベースとインターネット」参照)にあるかホモロジー検索(「1−3 ホモロジー検索」参照)やモチーフ検索(「2−2 配列モチーフと機能分類」参照)をすることが基本であり、類似配列が全くなかったり、類似配列があってもそれ自体が機能未知であれば、機能推定は行えないことになる。過去に実例がなければ現在の情報処理技術も全く無力であり、酵母の 6,000 の遺伝子の 1/3 は機能未知のままとなっている [4]。
ヒューマンゲノム計画の大きな目的の1つは病気の遺伝子を明らかにすることである。ここでも、ポジショナルクローニング(「1−1 ポジショナルクローニング」参照)と呼ばれるゲノム解析的アプローチがとられるようになった。遺伝病家系のリンケージ解析により、例えばある病気と関連した遺伝子が染色体上の特定の位置にあるらしいことが分かれば、その部分のクローニング(ポジショナルクローニング)と塩基配列決定を行い、上記と同様に遺伝子領域を予測して機能推定を行うのである。
このように、実験的に配列決定さえ行えば、情報処理技術で機能についての手がかりを得ることができる。ところで、ゲノム解析が直接対象とするのは「一次元的なかたち」であるが、実際にはDNAもタンパク質も「立体的なかたち」をした分子である。タンパク質の立体構造はその特異的な機能と深い関係がある(「2−3 球状タンパク質の分類」「2−4 膜タンパク質の分類」参照)ので、配列だけでは機能を推定できないタンパク質も、立体構造を決めることができれば、機能についての手がかりを得ることができるだろう。近年、タンパク質の立体構造を決定するX線結晶解析やNMRなどの実験技術は飛躍的に進歩し、これに伴い立体構造既知のタンパク質の数も急増している。しかしながら、発想としては依然として遺伝子クローニングと同様であり、まず特定の機能と特定のタンパク質に興味の対象があり、機能についてのより深い知見を得るために、タンパク質の立体構造決定が行われている。酵母の全タンパク質の立体構造をシステマティックに決めてしまおうといったゲノム解析的な発想への転換は、まだ行われていない。
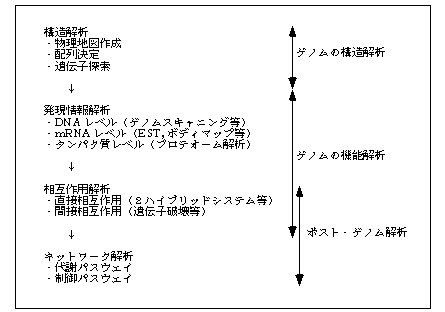
システマティックなシーケンシングの次のステップとしてすでに行われているのが、遺伝子の発現情報をシステマティックに解析することである。これには様々な方法があり得るが、ゲノム DNA 上でプロモーター領域などを調べる DNA レベルの解析、特定の cDNA ライブラリーから遺伝子発現情報を決める mRNA レベルの解析、さらにタンパク質レベルの解析と、3つのレベルに分けて考えることができる。タンパク質レベルの解析では、ある場所で発現している全タンパク質の二次元電気泳動といった解析が以前より行われていたが、最近はゲノム解析の次のステップとしてこれをプロテオーム解析と呼ぶことがある(「2−1 二次元電気泳動によるスキャニング法」参照)。遺伝子(gene)全体がゲノム(genome)であるのに対して、タンパク質(protein)全体がプロテオーム(proteome)である。
cDNA レベルの解析では、ライブラリーからできるだけ多くの cDNA をとって区別するために、EST (Expressed Sequence Tag) と呼ぶ配列断片情報のタグをつけることが盛んに行われており、さらにこれに発現場所、発現量、その他の情報が付加されたものをボディマップと呼ぶ。
ゲノム DNA レベルの解析はゲノムスキャニングなど、実験技術の面で今後大きく発展するものと思われる(「二次元電気泳動によるスキャニング法」参照)。
遺伝子がもつ配列情報と発現情報がすべて分かれば、ゲノム解析は終了するのだろうか。遺伝子が生命の基本部品であるとの言い方をすると、ここまでのゲノム解析は部品を個別に解析するアプローチである。しかしながら、個々の部品が分かっても生命のシステム全体が分かるわけではない。部品がどのように組合わさってシステムが構成されているかが解明されなければ、生命現象を理解することはできないのである。そこで非常に重要な概念となるのが、部品間のつながり、すなわち「分子間相互作用」である。
すでに酵母など全塩基配列決定が終了した生物種では、システマティックシーケンシングの時代からシステマティック機能解析の時代へと移りつつある。そして今後の機能解析の中心は、分子間相互作用のシステマティックな解析(「3−1 タンパク質相互作用の系統的解析を目指して」参照)となるだろう。これは大きく分けると生化学的な方法で分子間の直接的な相互作用を調べることと、遺伝学的な方法により分子間の間接的な相互作用(遺伝子間相互作用)を調べることに分けられる。このようなアプローチをゲノム計画の延長とみるか、ゲノム計画終了後の新しいプロジェクトとみるかは議論のあるところだが、本書では図1に示したように、これはポスト・ゲノム計画と位置づけている。分子間相互作用、すなわち生命系を構成する部品と部品のつながりを系統的に調べることができれば、そこから生命のシステム配線図を書き上げることも夢ではない。
| データベース* | データ解析 | |
|---|---|---|
| ゲノム解析における 情報処理 |
分子の構造データベース
・塩基配列(GenBank, EMBL, DDBJ) ・アミノ酸配列(SwissProt, PIR, PRF) ・立体構造(PDB) 分子の機能データベース ・プロモータ(EPD) ・転写因子(Transfac) ・タンパク質モチーフ(Prosite) |
分子の構造解析
・ホモロジー検索 ・立体構造検索 ・立体構造予測 分子の機能解析 ・モチーフ抽出 ・モチーフ検索 ・機能予測 |
| ポスト・ゲノム解析 における情報処理 |
分子間相互作用データベース
・酵素反応(LIGAND) ・情報伝達(BRITE) パスウェイデータベース ・代謝系(PATHWAY) ・制御系 | 分子間相互作用の解析
・相互作用検索 ・相互作用予測 ・パスウェイ構築 パスウェイの解析 ・パスウェイ比較 ・パスウェイ工学 |
ヒューマンゲノム計画は、別の側面でも新しい生物学の流れを作りつつある。それは生物学の情報化であり、実験技術と情報処理技術の融合、あるいは生物学と情報科学の融合である [5-8]。ゲノム計画がもたらす大量データを処理するためにコンピュータが不可欠であることは言うまでもなく、さらに「かたち」から「はたらき」を予測するために、情報処理技術の有効性と重要性が広く認識されるようになった。もはや実験技術と同様に、情報処理技術の進歩がなければ、生物学の進歩はあり得ない。ただ、これまでの情報処理技術はそれほど高度な内容のものではなく、例えば機能予測にしても過去に前例があるかを調べているに過ぎなかった。しかしながら、ポスト・ゲノムの時代を迎えて、生物学の情報科学的な側面も、部品の解析からシステムの解析へと新しい発展をしつつある。
これまでのホモロジー解析やモチーフ解析では、新しい配列が既知の配列と類似しているか部品と部品の比較をしている。データベースに蓄積されているのは配列や立体構造など部品に関する「ファクト」(事実データ)であり、ファクトがあるかないかの「検索」が主な利用形態であると想定してデータベースが作られている。一方、データベースに相互作用のような「関係」が蓄積されていると、それを「演繹」して新しい関係を導き出すことができる。例えば、酵素遺伝子は基質と生成物の関係を表現しているので、新しく決定されたゲノムの酵素遺伝子を集めて既知の代謝システムが再構成できるかどうかを調べることができる(「3−2 代謝系の演繹データベース」参照)。しかしながら、既知の生物学的知識はこのように利用できる形にはほとんどコンピュータ化されていない。知識の利用とは単に蓄積された知識を検索するだけでなく、知識と知識を組み合わせて推論することである。生物知識情報のコンピュータ化と推論過程のコンピュータ化(「3−3 細胞内局化の知識ベースシステム」参照)を行うことにより、部品と部品の比較だけでなく、部品からシステムの構築、あるいはシステムとシステムの比較といったポスト・ゲノム時代の情報処理(表1)が可能となっていくだろう。
生物学では、これまで個々の生物学者の興味の対象として断片的な知識しか得られていなかったために、物理学や化学のように経験的知識が未だ原理として体系化されていない。ヒューマンゲノム計画により、生物学の歴史の中で初めてシステマティックにデータを得る技術が確立し、生物の「すべて」を調べることができるようになってきた。生物学のデータも秩序とゆらぎ、あるいは空間と時間といった物理学的な視点で眺め(「3−4 進化のダイナミックス」参照)、そこから生命現象の基本原理解明への道が開けるだろう。以下では、ヒューマンゲノム計画がもたらしつつある新しい生物学、すなわちニューバイオフィジックスに対する私見を述べる。
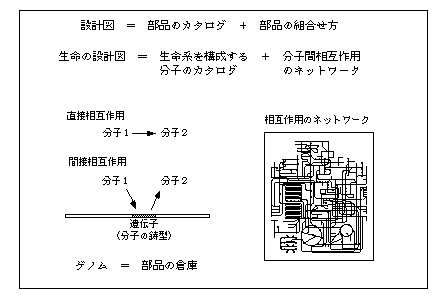
ヒューマンゲノム計画が発足した当時、これは生命の設計図を読みとる計画であると言われていた。では、酵母ゲノムの全塩基配列はすでに読みとられ、コンピュータにすべて入っているのだから、コンピュータの中で酵母のライフサイクルをシミュレートできるのだろうか。もちろん現状ではそのようなことは全く実現性のない話である。ゲノムは設計図であるとする立場からは、これは我々がまだ設計図を読みとる能力がないからであると主張するかもしれない。しかし、別の立場からはゲノムは設計図などではなく、ただ単に生命が必要とする部品の倉庫に過ぎないと考えることもできる。すなわち、本当の設計図には部品をいつどこでどのように組み合わせていくかが記載されているべきで、ゲノムに書かれているのは部品の鋳型の情報(遺伝子コード領域)とその情報の取り出し方(遺伝子発現制御領域)だけなのである。部品の組合せ方の情報は分子間相互作用が形成するネットワークにあり、これをすべて明らかにして生命のシステム回路図のようなものができたときに、生命の設計図を読みとったと言えるのではないだろうか(図2)。
| テクノロジー | 内容 |
|---|---|
| 遺伝子工学 | DNA の組換え |
| タンパク質工学 | 変異タンパク質による立体構造改変 |
| 進化分子工学 | RNA の実験室内進化 |
| パスウェイ工学 | 生体内パスウェイ情報に基づく低分子の設計 |
物理学では物質とその相互作用の概念がいずれも重要であるのに対し、分子生物学では個々の分子に対する興味が中心で、分子間相互作用という観点はあまり重視されていない。例えば、このタンパク質はDNA結合タンパク質である、あるいはプロテインキナーゼであるといった具合に、分子単位に物事を考える場合が多いが、タンパク質の機能とは当然相手となる分子があって成り立つわけで、どの分子とどの分子がどんな相互作用をしているといった具合に、分子間相互作用を単位とした見方が必要である。これに関連して、構造予測と機能予測の問題について考えてみよう。
アミノ酸配列からタンパク質の立体構造を予測する問題は、生物物理学の大問題として長年研究が行われている。それは、タンパク質の立体構造と機能の間には深い関連があり、機能を知るためには立体構造を知らなければならないからだとされている。ところで、ここで言う立体構造には2つの意味があることに注意しなければならない。
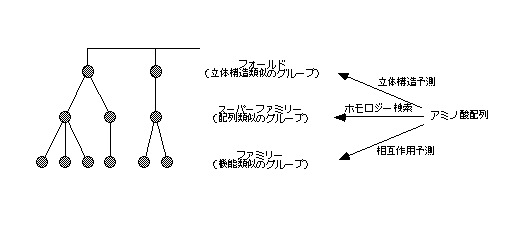
構造予測の場合の立体構造はポリペプチド鎖の全体的な折れ畳み(フォールド)を問題にするが、機能との関連は機能部位の局所的な立体構造についての話である。そして、機能は非常に特異的であり、配列の類似性はあるのに機能的には異なるタンパク質は数多く存在する。一方、全体的なフォールドは機能グループよりも配列グループよりも非特異的であり、類似配列であればフォールドも同じであるが、類似配列でなくてもフォールドは同じになり得る(図3)。従って、機能未知のタンパク質が現在の立体構造予測法(囲み記事参照)により特定のフォールドへ帰属されたとしても、配列から機能予測を直接行った以上に、機能についての手がかりが得られるわけではない。
物質がもつ粒子と波動の二面性は量子力学で見事に融合した。一見相反する事象には、それを包含する新しい概念や理論の可能性が秘められている。生物学の問題にも、とくに生命の起源と生物種の進化の問題には、一見相反する解釈をめぐって多くの論争がある。例えば、系統樹の問題を考えてみよう。生物界は以前は原核生物(Prokaryotes)と真核生物(Eukaryotes)の2つに分けられていたが、1970年代に原核生物は真性細菌(Eubacteria)と古細菌(Archaebacteria)に区別されるようになった。古細菌とはメタン生成細菌、耐熱菌を始め原始地球を反映したかのような極限環境で生きている細菌である。そして3者の系統関係として、古細菌は真性細菌より真核生物に近いとする系統樹が有力視されている。このような系統樹を書くには、オーソロガス遺伝子、すなわち種分岐以前から存在した共通の遺伝子を時計として、その変化から推定する。これが分子進化であるが、形態などマクロな観点から作られた系統樹と必ずしも合わないのは当然にしても、用いた分子によって系統樹が異なる場合がよくある。
生物のデータは何となく曖昧で非決定的なイメージがあるにもかかわらず、系統樹が表現する生物種の分類関係は決定的である。ところで、1つの生物個体が1つのゲノム塩基配列で定義できるとすると、1つの生物種は変異を伴ったゲノム塩基配列のポピュレーションである(「3−4 進化のダイナミックス」参照)。すなわち地球上に生存するすべての生物個体をゲノム塩基配列空間の1点に対応させるとすると、生物種は局所的な点のクラスターに相当することになる。そして、ゲノム塩基配列空間を段階的に平滑化し、より低い解像度で眺めたときのより大きなクラスターが、階層的な生物分類のグループに対応することになる。さらに、生物の進化とは、ゲノム塩基配列空間の点集合が、宇宙の時間軸とともに変化してきたことである。生物の分類と進化をこのように眺めると、どんな平滑化とグループ化を行ったかで、たとえば、真核生物、真性細菌、古細菌の系統関係、あるいは動物、植物、菌類の系統関係に関して、一見相反する結果が出ることも、本質的には矛盾していないのかもしれない。
生命の起源についても、全くの偶然に一度だけ起こった現象として生命が誕生したのだろうか、それともいろいろな場所でいろいろな時代に何度も生命が誕生し、その中で限られたものが残って現在の生物界へと進化したのだろうか。生命の誕生とその進化は地球の誕生と進化、さらには地球をとりまく宇宙の進化とともに考えていかなけらばならない問題である。地球上に現存する生物のゲノム情報をすべて読みとることにより、また過去の生物のゲノム情報も部分的に読みとることにより、生命の起源と進化、生物の多様性と分類といった生命科学の基本問題に対する手がかりが得られるだろう。
本稿で述べてきたように、ヒューマンゲノム計画がもたらす新しい生物学では、コンピュータを武器とする情報科学的側面だけでなく、理論的考察を武器とする物理学的側面が生まれつつある。理論物理学が自然界の神秘を解き明かしてきたように、ゲノム情報に基づく理論的考察は生物界の神秘を解き明かしていく。自然界に対する理論と生物界に対する理論の融合が、物理学と生物学の融合をもたらす。これがニューバイオフィジックスではないだろうか。
|
発想の転換 1.逆転写 遺伝情報はDNAからRNAへ転写され、さらにRNAからタンパク質へ翻訳されて発現する。これがクリックが提唱した分子生物学のセントラルドグマである。ところが、一部のウイルス(レトロウイルス)ではRNAを鋳型にしたDNA合成、すなわち逆転写が行われていることが発見された。これは、その後のRNAの役割に関する発想の転換の契機となったのである。セントラルドグマでは脇役的存在でしかなかったRNAが、実は生命の初期にはDNAもタンパク質も存在しないRNAの世界を形成していたとする考え方が、今では有力となっている。
2.逆遺伝学
3.逆フォールディング
|
オーソロガス遺伝子とパラロガス遺伝子 遺伝子または遺伝子産物(タンパク質、RNA)における配列の類似性は機能的関連を示し、一般にホモロジー(homology)と呼ばれている。分子進化の観点から、ホモロジーはオーソロジー(orthology)とパラロジー(paralogy)に区別して考えることができる。オーソロジーとは種分岐により生じた遺伝子間の関連で、例えばヒトのヘモグロビンα鎖とウマのヘモグロビンα鎖の遺伝子は、オーソロガス遺伝子である。一方、パラロジーは遺伝子重複により生じた遺伝子間の関連で、例えばヘモグロビンα鎖とヘモグロビンβ鎖の遺伝子はパラロガス遺伝子である。遺伝子重複の後に種分岐したヒトのヘモグロビンα鎖とウマのヘモグロビンβ鎖のようなパラロガス遺伝子もあれば、恐らく種固有の機能に関連した、種内に限られた遺伝子重複によるパラロガス遺伝子もある。 |
文献